

What is Single-Cell ATAC Sequencing (scATAC-seq)?
Single-cell ATAC-seq (Assay for Transposase-Accessible Chromatin using sequencing) is an innovative technique that enables the investigation of chromatin accessibility at the single-cell level. The accessibility of chromatin—whether it is in an open or closed configuration—critically impacts gene expression and cell fate. Therefore, understanding chromatin openness is vital for probing gene regulation, cell type specificity, and underlying disease mechanisms.
ATAC-seq uses the 'cut-and-paste' action of the Tn5 enzyme. This enzyme adds short DNA fragments with sequencing tags to open chromatin areas, marking them as accessible. ScATAC-seq advances this concept by applying it to individual cells, allowing the capture of unique chromatin accessibility profiles for each cell. This approach offers new perspectives on cellular heterogeneity, developmental processes, and disease pathophysiology.
Unlike other genomic techniques such as RNA-seq, scATAC-seq is specifically attuned to DNA fragments within the cell nucleus. It primarily focuses on examining the architecture of chromatin and the exposure of regulatory regions. Thus, it elucidates the status of gene regulatory elements within cells, including promoters and enhancers, rather than merely measuring transcript abundance.
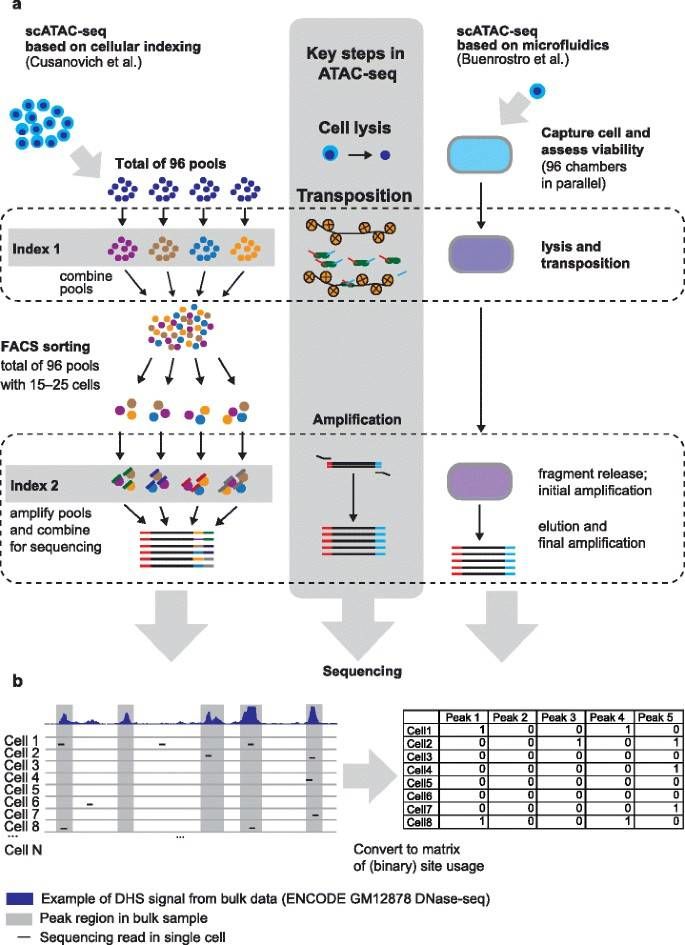
Single-cell assay for transposase-accessible chromatin (scATAC-seq) methods. (Pott, S., et al., Genome Biol, 2015)
The Rise of Single-Cell ATAC-Seq Assays
Single-cell ATAC-seq was developed to better understand chromatin accessibility at the single-cell level, which has greatly improved our understanding of gene regulation. As technology advanced, scATAC-seq evolved from early low-throughput techniques to the contemporary high-throughput methodologies, establishing itself as a pivotal tool in the field of epigenetics.
1. Early Developments in Single-Cell ATAC-Seq
Single-cell ATAC-seq was first introduced in 2015, with pioneering contributions from Shendure and Greenleaf laboratories, each proposing distinct approaches for chromatin accessibility analysis at the single-cell level. Shendure's method employed a double indexing strategy using a transposase to attach unique barcodes to each cell nucleus, allowing for the differentiation of ATAC-seq data at the individual cell level. Despite its ability to process large numbers of cells, the method suffered from limited resolution due to a low read count per cell, usually below 3000. Conversely, Greenleaf's method utilized microchamber-based physical separation and microscope positioning for precise single-cell capture, achieving higher data quality with average reads per cell reaching up to 73,000. However, this approach was hindered by its complexity, low throughput, and limited cell numbers.
2. Breakthrough and Popularization by 10x Genomics
In 2018, the introduction of single-cell ATAC-seq technology by 10x Genomics marked a significant advancement and set a new standard for widespread application. This technology, akin to their single-cell RNA-seq solution, entails cell encapsulation in gel beads followed by ATAC-seq library construction. Unlike RNA-seq, scATAC-seq gel beads do not contain molecular barcodes since the measurement is centered around DNA fragments within the cell nucleus rather than transcripts. Importantly, scATAC-seq enables direct sequencing of open chromatin regions without fragment interruption, yielding expansive epigenetic information. This evolution significantly enhanced both the throughput and accuracy of data, cementing scATAC-seq as a staple technique in epigenomic research.
3. Innovations in Spatial ATAC-Seq
Progress in technology has propelled ATAC-seq applications from single-cell resolution to the spatial dimension, giving rise to Spatial-ATAC-seq. This innovative approach, notably advanced in 2021, integrates ATAC-seq with tissue sectioning techniques, allowing researchers to decipher chromatin accessibility within the spatial context of tissues. By offering concurrent information on cell-specific chromatin states and their spatial dynamics within tissues, Spatial-ATAC-seq facilitates comprehensive insights into cellular functions and their regional interactions. This represents a significant leap, steering epigenomic research into the realm of spatial genomics.
4. Challenges and Future Prospects of Technological Advancement
While single-cell ATAC-seq has achieved substantial progress, it confronts ongoing challenges. Compared to bulk ATAC-seq, single-cell ATAC-seq has limitations in capturing open chromatin regions. It also faces challenges from noise and data complexity, making analysis and interpretation harder. Additionally, accurate cell type identification frequently necessitates integration with single-cell RNA-seq data, representing a significant challenge in its application. However, continuous technological optimization and the emergence of more sophisticated computational tools are likely to expand the applicability and enhance the resolution of scATAC-seq data interpretation.
Overall, single-cell ATAC-seq has evolved from basic low-throughput methods to highly precise, high-throughput systems. Its widespread adoption is advancing the fields of epigenetics, cellular biology, and disease research, promising a future of expansive potential.
How scATAC-seq Works: A Step-by-Step Guide
The scATAC-seq leverages the "cut-and-paste" activity of the Tn5 transposase to insert DNA fragments, equipped with sequencing adapters, into regions of open chromatin. This method offers a powerful approach to understanding chromatin accessibility at the single-cell level. Below are the detailed experimental steps employed in scATAC-seq:
Overview:
- Start with a suspension of isolated cell nuclei.
- Introduce Tn5 transposase to facilitate fragmentation, where the enzyme enters intact nuclei to cleave open chromatin regions, simultaneously inserting 10x barcodes into the DNA fragments.
- Single nuclei are then encapsulated into droplets using the 10x Chromium system. Subsequently, the tagmented DNA fragments undergo single-cell barcode tagging through Next GEM technology, allowing all fragments from an individual cell to share a unique barcode.
- Finally, these fragments are PCR amplified and sequenced. Data analysis maps the sequencing reads and traces each read back to its cell of origin via the barcode.
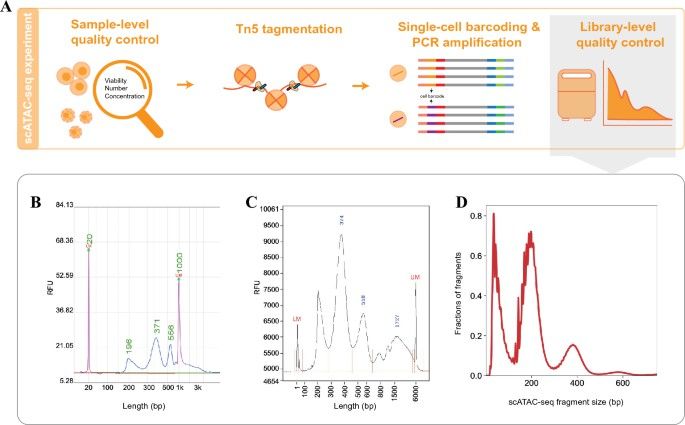
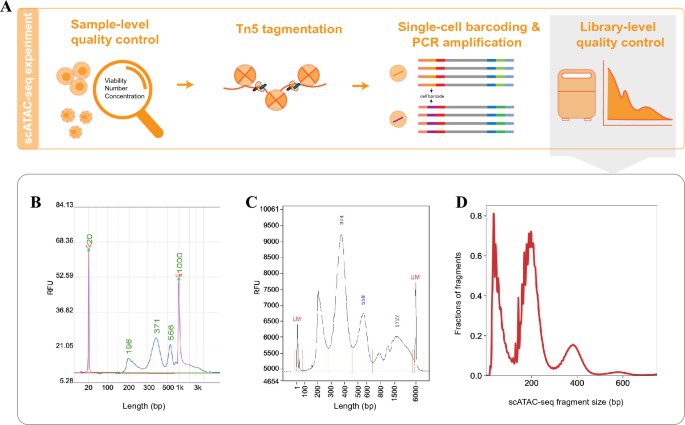
General steps and quality control of a conventional scATAC-seq experiment. (Shi, P., et al., aBIOTECH,2022)
Detailed Steps
Step 1: Nuclear Isolation
scATAC-seq starts with a suspension of cell nuclei to ensure accurate labeling. This can be prepared from fresh, frozen, or cryopreserved cells and tissues using specific kits and protocols.
Step 2: Tagmentation
Isolated nuclei are subjected to extensive tagmentation by introducing the Tn5 transposase protein. In scATAC-seq, tagmentation involves attaching a 10x Genomics barcode—serving as a unique identifier—across all accessible chromatin regions. These barcodes allow DNA fragments to adhere to 10x primers in the droplets formed in step 3.
This critical step is driven by Tn5 transposase, central to ATAC-seq assays. Tn5 is a bacterial enzyme that can penetrate open chromatin and integrate DNA fragments within the host genome. It operates through a "cut-and-paste" mechanism, tailored here for ATAC-seq. To enhance its functionality, scientists have modified Tn5, loading it with a recognizable 19-base pair "tag," fostering its widespread application in biomedical research.
Step 3: Single-Cell Barcoding
The 10x Chromium X instrument, employing microfluidic technology, augments each tagmented DNA fragment with a cell-specific barcode. The 10x Genomics methodology utilizes GEMs—water-in-oil emulsion droplets. Each GEM encapsulates a single nucleus with a barcode-laden gel bead, ensuring that all tagmented DNA fragments from the same cell share the same barcode.
Barcode incorporation into the sequencing index facilitates subsequent library construction and quality control measures.
Step 4: Sequencing
The barcoded sequenced indices, after amplification, undergo sequencing via next-generation sequencing technologies.
Step 5: Data Analysis
scATAC-seq data analysis aims to identify regions of open chromatin across the genome. A cornerstone of this analysis is peak calling, wherein specialized algorithms like 10x Genomics' CellRanger and MACS2 pinpoint areas in the genome where sequencing reads are densely clustered, corresponding to accessible chromatin regions. Peak calling can be performed pre- and post-data filtration to eliminate low-quality cells, repetitive reads, and peak-associated noise.
With single-cell barcodes, algorithms allocate peaks back to the cells of origin. Subsequently, cell clustering derived from 10x single-cell ATAC data facilitates the identification of all cell types present in a sample. Delving into the chromatin accessibility characteristics of clusters, such as evaluating known cell type markers, permits the annotation of cell types for each cluster.
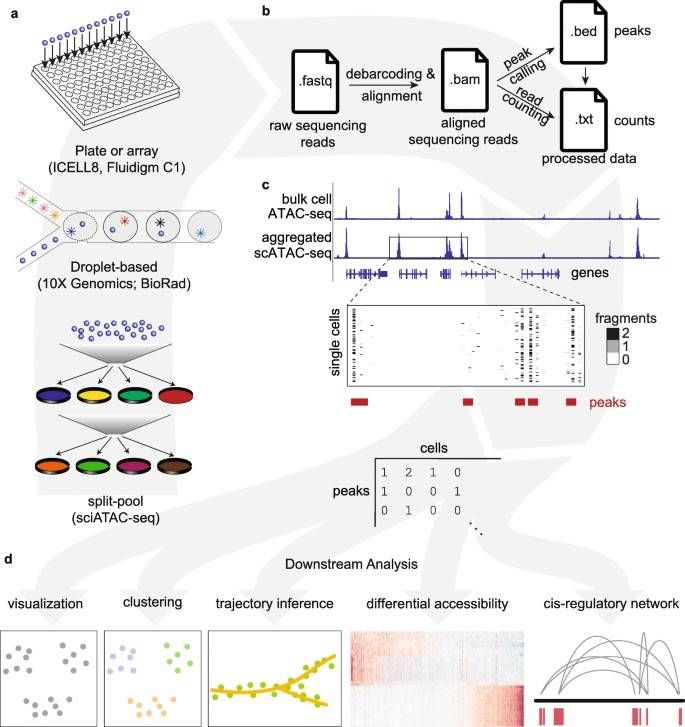
Schematic overview of single-cell ATAC-seq assays and analysis steps. (Chen, H., et al., Genome Biol, 2019)
Further analysis can include conducting cell clustering prior to peak calling within each cluster, enabling insights into rare cell accessibility features. Following this, exploration into transcription factor motif enrichment and constructing transcription factor regulatory networks or interaction analyses can be undertaken.
Advantages and Limitations of scATAC-Seq
Advantages of scATAC-Seq
- Single-Cell Resolution: Unlike traditional bulk ATAC-seq, scATAC-seq enables the assessment of chromatin accessibility at an individual cell level, revealing unique features of each cell. This high-resolution technique uncovers tissue heterogeneity, tracks different cellular states, and identifies rare cell populations, providing crucial insights into cellular function and transcriptional regulation and aiding in understanding complex biological systems.
- Accurate Cell State and Lineage Identification: By analyzing chromatin accessibility patterns, scATAC-seq allows scientists to pinpoint cell states with precision. This is particularly beneficial for studying developmental processes, cellular differentiation, and cell state changes during pathological events, offering comprehensive lineage information and identifying regulatory elements that are challenging to detect with conventional methods.
- High Throughput: Modern scATAC-seq technologies can process data from thousands of cells at once, improving speed and depth. This makes it easier to analyze many cell types and states, helping to speed up research and create large-scale cell atlases.
- Identification of Cell Type-Specific Regulatory Elements: By comparing chromatin accessibility across various cell types, scATAC-seq identifies regulatory elements specific to each cell type. This helps elucidate the functionality of gene regulatory regions, thereby deepening our understanding of cell fate decisions and functional determination.
- Dynamic Tracking Capabilities: scATAC-seq is excellent for studying changes in chromatin structure during biological processes. Researchers can track how cells respond to stimuli or treatments over time, which helps in understanding disease progression, immune responses, and how drugs work.
- Independence from RNA Information: In contrast to RNA-seq, scATAC-seq does not depend on RNA abundance, circumventing issues related to RNA degradation or abundance variability. It directly measures the open state of DNA regions within the nucleus, which is particularly important for studying gene regulation.
- Integration with Other Omics Data: scATAC-seq data can be integrated with data from other single-cell technologies, such as scRNA-seq, to offer a comprehensive view of chromatin and gene expression dynamics, thus unravelling the complexity of gene regulatory networks.
Limitations of scATAC-Seq
- Data Processing and Interpretation Challenges: scATAC-seq generates vast amounts of data with high noise, necessitating sophisticated algorithms and tools for analysis, like data preprocessing, peak calling, and cell type annotation. These analyses require substantial computational resources, expertise, and time, which can impose technical and financial burdens on researchers.
- Low Signal Intensity and High Technical Variability: The relatively small amount of sequencing data obtained from individual cells may lead to low signal intensity. Additionally, technical variability can introduce errors, affecting analytical outcomes. Ensuring sufficient labeling and adequate sequencing depth presents a technical challenge that demands specialized equipment and meticulous experimental design.
- Cost and Experimental Complexity: Due to its high resolution and throughput, scATAC-seq requires costly reagents, complex machinery, and detailed experimental procedures, potentially limiting its application in resource-constrained laboratories. The intricate experimental workflow and high skill demand for technicians add to the operational complexity and cost.
- Limited Capture of Rare Cell Populations: Although scATAC-seq has advantages in capturing rare cells, detecting extremely rare or sparsely distributed cells remains challenging. The information obtained from individual cells may not fully represent the complexity of the entire rare cell population, necessitating the use of complementary methods.
- Sample Type Limitations: scATAC-seq faces challenges when processing certain sample types, such as hard tissues or low-quality cells, possibly due to obstacles encountered during sample preparation or lysis. The texture and condition of samples can affect the success rate of experiments and data quality.
- Chromatin Capture Capacity: In some cases, particularly for low-abundance or tightly closed chromatin regions, the detection capability of scATAC-seq may be inferior to that of bulk ATAC-seq. Consequently, critical regulatory regions could be overlooked or inadequately characterized.
- Defining Cell Types: While scATAC-seq can reveal chromatin accessibility, identifying specific cell types and subtypes often requires additional techniques like scRNA-seq, increasing experimental complexity and difficulty in data integration.
- Complex Data Handling: Analyzing the extensive data generated by scATAC-seq involves sophisticated computational skills, especially during chromatin accessibility and peak annotation, requiring a high level of bioinformatics expertise from researchers.
Applications of scATAC-seq
This section outlines the diverse applications of scATAC-seq in unveiling cellular heterogeneity, monitoring disease dynamics, identifying gene regulatory elements, and integrating epigenetics with gene expression. These insights underscore the method's expansive potential in both fundamental and clinical research.
Revealing Cellular Heterogeneity
scATAC-seq excels in distinguishing variations in chromatin accessibility within cell populations, thereby advancing the study of cellular heterogeneity. By examining chromatin openness, researchers can identify distinct cell types or subtypes, including elusive rare cell subpopulations. Unlike conventional marker-based identification methods, scATAC-seq does not rely on known cell markers, enabling the discovery of novel cell types or subtypes and offering fresh perspectives for disease research. For instance, in oncology, scATAC-seq facilitates the exploration of intratumoral heterogeneity, uncovering chromatin accessibility differences between tumor subpopulations, and aiding in the identification of cells associated with tumor progression, drug resistance, or metastasis.
